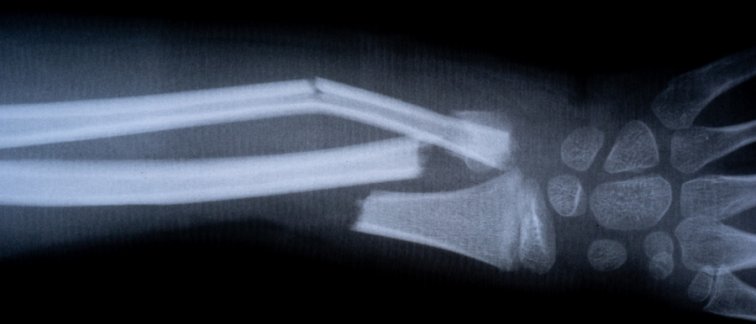
Osteogenesis Imperfecta patients can experience hundreds of bone fractures in their lifetime caused by defects in collagen type I.
Compared to genetic diseases caused by a specific mutation, it is caused by hundreds of different mutations in the collagen type I genes. The quesstion is how to treat effectively a disease with such high genetic heterogeneity?
The molecular culprit of the disease is collagen type I, the most abundant protein in the human body. Its presence in many anatomical locations explains the presentation of additional features such as dentinogenesis imperfecta, hearing loss, blue sclerae and cardiovascular problems.
Collagen type I is especially important in bones where it provides elastic strength; without collagen our bones would shatter after exposure to mechanical impact during physical activities.
In the vast majority of patients, Osteogenesis Imperfecta is caused by mutations in the COL1A1 or COL1A2 genes which produce collagen type I. Hundreds of different mutations have been identified which complicate the development of a uniform treatment strategy. These can be divided to mutations leading to less collagen production and mutations leading to collagen with abnormal structure.
Their common point is the bone fragility for which no effective therapy exists. Patients are largely treated with bisphosphonates which do not target the primary disease defect and may have side effects after long term use. This project aims the development of novel treatment for these patients. We aim to address the different types of collagen defects by using a combined approach of gene and pharmacological therapy.


